Мой файл с результатами Chip-seq экспериента называется chipseq_y14/chipseq_chunk5.fastq и находится по адресу /srv/databases/ngs/chipseq_y14 на диске P.
Для начала нужно провести контроль качества прочтений. Это можно сделать при помощи программы FastQC. Она проводит набор стандартных статистических анализов, которые можно использовать для предварительной оценки релевантности данных и выявления возможных проблем в полученной последовательности.
Результатом работы является сайт html и архив .zip. Я не вижу смысла приводить из здесь, так как самая главная для меня информация находится в двух картинках.
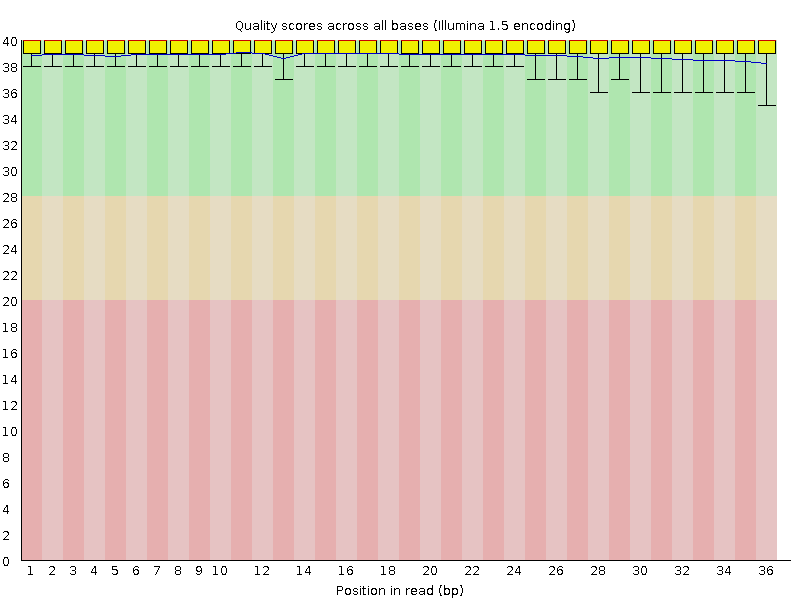
Оценка качества определения основания в каждой позиции:
Основная информация:
Прочтения имеют очень хорошее качество, так что нет смысла чистить их программой Trimmomatic.
Далее я картировал прочтения на геном человека при помощи алгоритма BWA-MEM.
Использованная команда: bwa mem path/to/hg19/GRCh37.p13.genome.fa chipseq_chunk5.fastq > chipseq_chunk5.sam
Так как уже дан проиндексированный геном, индексировать результат не нужно.
Анализ последовательностей проводился с помощью пакета samtools. Ниже указаны команды и их функция:
Файл с информацией о количестве чтений:chipseq_chunk5.idxstats
Видно, что основная масса прочтений (6185 из 6572 ) откартировалась на 12 хромосому.
Программа MACS осуществляет поиск пиков. Команда: macs2 callpeak --nomodel -n chip5 -t chipseq_chunk5.sorted.bam
В итоге было найдено 8 пиков шириной от 243 до 585 нуклеотидов, расположенных в одном регионе 4-й хромосомы.
Результатом выдачи являются 3 файла, в которых находится нужная информация о найденных пиках:
Все что нужно знать о найденных пиках:
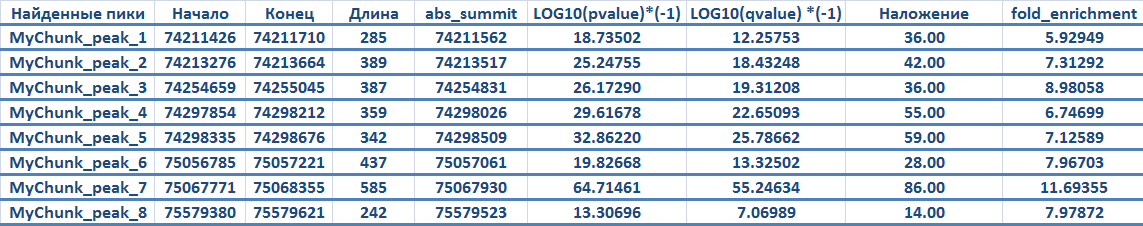
В таблице содержатся отрицательные десятичные логарифмы p-value и q-value, а значит трактовать их надо не так, как обычные значения. Здесь работает принцип "чем больше, тем лучше", поскольку при повышении этих значений p-value и q-value падают. По ним можно судить о достоверности пиков, и, согласно этому критерию, все находки оказались достаточно надежными
Мне не удалось визуализировать информацию из файла .narrowPeak, Genome Browser просто отказался принимать этот файл (разумеется я вписал в начало требуемые по инструкции строчки). Причину ошибки мне не удалось установить.
Зато я смог загрузить файл summits.bed (о такой возможности я также узнал из инструкции).
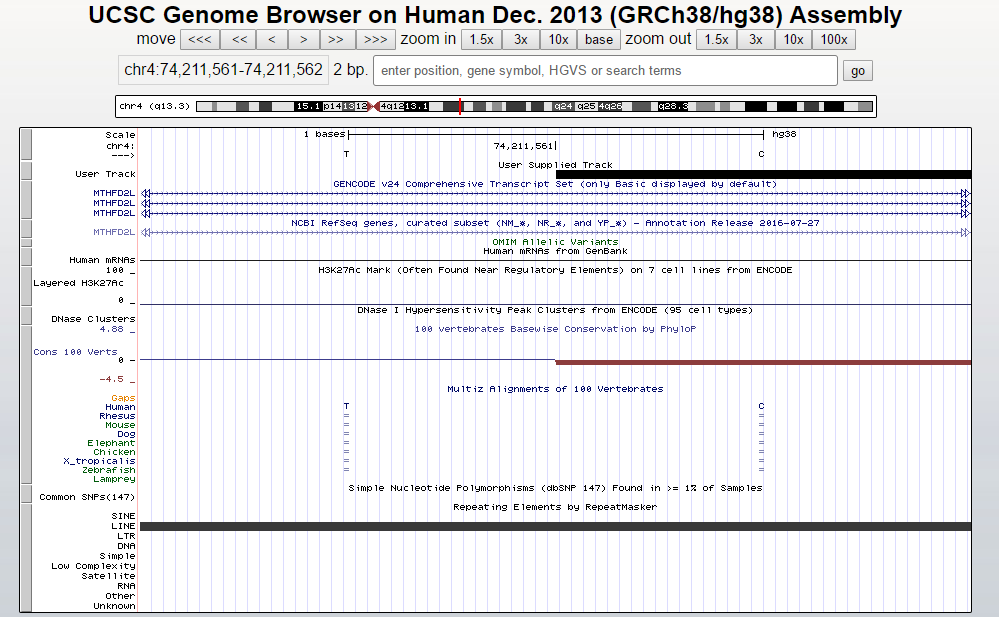
Я не получил никакой информации о пиках и их перекрываниях. Возможно, их просто нет. Или данные неточны. Или какая-то программа сработала не так как надо.
Конвертируем файл .narrowPeak в fasta-формат.
Для этого я применил команду /srv/databases/ngs/tools/seqtk/seqtk subseq /srv/databases/ngs/hg19/GRCh37.p13.genome.fa MyChunk_peaks.narrowPeak > results.fa
Результат: results.fa
После я построил выравнивание, используя программу JalView.

В итоге мне не удалось идентифицировать консенсусную последовательность (~ 4 - 8 nts), так как выравнивание получилось низкого качества.
© Борисов Евгений 2017